

地址:廣州市南沙區大崗鎮智新一路8號聯東U谷廣州南沙國際企業港7棟5樓
產品咨詢:13392693259
耗材電話:18998328368
售后服務:020-87684117
傳 真:020-87684846
Email:info@gdana.com
網址:http://www.yytqd.com.cn/
原標題:GB5009.11-2014食品安全國家標準 食品中總砷及無機砷的測定
1、范圍
本標準第一篇規定了食品中總砷的測定方法。本標準第二篇規定了食品中無機砷含量測定的液相色譜-原子熒光光譜法、液相色譜-電感耦合等離子體質譜法。
本標準第一篇第一法、第二法和第三法適用于各類食品中總砷的測定。第二篇適用于稻米、水產動物、嬰幼兒谷類輔助食品、嬰幼兒罐裝輔助食品中無杋砷(包括砷酸鹽和亞砷酸鹽)含量的測定。
第一篇總砷的測定
第一法電感耦合等離子體質譜法
2、原理
樣品經酸消解處理為樣品溶液,樣品溶液經霧化由載氣送入ICP炬管中,經過蒸發、解離、原子化和離子化等過程,轉化為帶電荷的離子,經離子采集系統進入質譜儀,質譜儀根據質荷比進行分離。對于一定的質荷比,質譜的信號強度與進入質譜儀的離子數成正比,即樣品濃度與質譜信號強度成正比通過測量質譜的信號強度對試樣溶液中的砷元素進行測定。
3、試劑和材料
注:除非另有說明,本方法所用試劑均為優級純,水為GB/T6682規定的一級水
3.1試劑
3.1.1硝酸(HNO3):MOS級(電子工業專用高純化學品)、BV(Ⅲ)級
3.1.2過氧化氫(H2O2)。
3.1.3質譜調諧液:Li、Y、Ce、Ti、Co,推薦使用濃度為10ng/ml
3.1.4內標儲備液:Ge,濃度為100g/mL
3.1.5氫氧化鈉(NaOH)。
3.2試劑配制
3.2.1硝酸溶液(2+98):量取20mL硝酸,緩緩倒入980mL水中,混勻。
3.2.2內標溶液Ge或Y(1.0g/mL):取1.0mL內標溶液,用硝酸溶液(2十98)稀釋并定容至
3.2.3氫氧化鈉溶液(100g/L):稱取10.0g氫氧化鈉,用水溶解和定容至100mL。
3.3標準品
氧化二砷(As2O3)標準品:純度≥99.5%。GB5009.11-2014
3.4標準溶液配制
3.4.1砷標準儲備液(100mg/L,按As計):準確稱取于100℃干燥2h的三氧化二砷0.0132g,加1mL氫氧化鈉溶液(100g/L)和少量水溶解,轉入100mL容量瓶中,加入適量鹽酸調整其酸度近中性用水稀釋至刻度。4℃避光保存,保存期一年。或購買經國家認證并授予標準物質證書的標準溶液物質。
3.4.2砷標準使用液(1.00mg/L,按As計):準確吸取1.00mL砷標準儲備液(100mg/L)于100mL容量瓶中,用硝酸溶液(2+98)稀釋定容至刻度。現用現配。
4、儀器和設備
玻璃器皿及聚四氟乙烯消解內罐均需以硝酸溶液(1+4)浸泡24h,用水反復沖洗,最后用去離子水沖洗干凈
4.1電感耦合等離子體質譜儀(icp-ms)
4.2微波消解系統。
4.3壓力消解器。
4.4恒溫干燥箱(50℃~300℃
4.5控溫電熱板(50℃~200℃
4.6超聲水浴箱
4.7天平:感量為0.1mg和1mg
5、分析步驟
5.1試樣預處理
5.1.1在采樣和制備過程中,應注意不使試樣污染。
5.1.2糧食、豆類等樣品去雜物后粉碎均勻,裝入潔凈聚乙烯瓶中,密封保存備用。
5.1.3蔬菜、水果、魚類、肉類及蛋類等新鮮樣品,洗凈晾干,取可食部分勻漿,裝人潔浄聚乙烯瓶中,密封,于4℃冰箱冷藏備用。
5.2試樣消解
5.2.1微波消解法
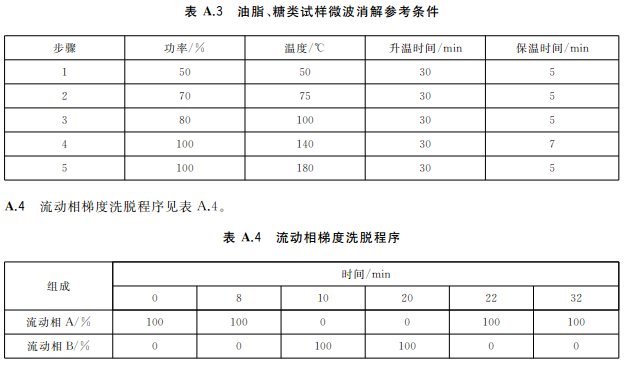
蔬菜、水果等含水分高的樣品,稱取2.0g~4.0g(精確至0.001g)樣品于消解罐中,加入5mL硝酸,放置30min;糧食、肉類、魚類等樣品,稱取0.2g~0.5g(精確至0.001g)樣品于消解罐中,加入5mL硝酸,放置30min,蓋好安全閥,將消解罐放入微波消解系統中,根據不同類型的樣品,設置適宜的微波消解程序(見表A.1~表A.3),按相關步驟進行消解,消解完全后趕酸,將消化液轉移至25mL容量瓶或比色管中,用少量水洗滌內罐3次,合并洗滌液并定容至刻度,混勻。同時作空白試驗。
5.2.2高壓密閉消解法
稱取固體試樣0.20g~1.0g(精確至0.001g),濕樣1.0g~5.0g(精確至0.001g)或取液體試樣2.00mL~5.00mL于消解內罐中,加入5mL硝酸浸泡過夜。蓋好內蓋,旋緊不銹鋼外套,放入恒溫干燥箱,140℃~160℃保持3h~4h,自然冷卻至室溫,然后緩慢旋松不銹鋼外套,將消解內罐取岀,用少量水沖洗內蓋,放在控溫電熱板上于120℃趕去棕色氣體。取出消解內罐,將消化液轉移至25mL容量瓶或比色管中,用少量水洗滌內罐3次,合并洗滌液并定容至刻度,混勻。同時作空白試驗。
5.3儀器參考條件
RF功率1550W;載氣流速1.14L/min;采樣深度7mm;霧化室溫度2℃C;Ni采樣錐,Ni截取錐。
質譜干擾主要來源于同量異位素、多原子、雙電荷離子等,可采用最優化儀器條件、干擾校正方程校正或采用碰撞池、動態反應池技術方法消除干擾。砷的干擾校正方程為:As=75As-7M(3.127)+82M(2.733)-83M(2.757);采用內標校正、稀釋樣品等方法校正非質譜干擾。砷的m/z為75,選Ge為內標元素。
推薦使用碰撞/反應池技術,在沒有碰撞/反應池技術的情況下使用干擾方程消除干擾的影響。
5.4標準曲線的制作
吸取適量砷標準使用液(1.00mg/L),用硝酸溶液(2+98)配制砷濃度分別為0.00ng/ml、1.0ng/mL、5.0ng/mL、10ng/ml、50ng/mL和100ng/mL.的標準系列溶液。
當儀器真空度達到要求時,用調諧液調整儀器靈敏度、氧化物、雙電荷、分辨率等各項指標,當儀器各項指標達到測定要求,編輯測定方法、選擇相關消除干擾方法,引內標,觀測內標靈敏度、脈沖與模擬模式的線性擬合,符合要求后,將標準系列引入儀器。進行相關數據處理,繪制標準曲線、計算回歸方程。
5.5試樣溶液的測定
相同條件下,將試劑空白、樣品溶液分別引入儀器進行測定。根據回歸方程計算出樣品中砷元素的濃度。
6、分析結果的表述

7、精密度
在重復性條件下獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%。
8、其他
稱樣量為1g,定容體積為25mL時,方法檢出限為0.003mg/kg,方法定量限為0.010mg/kg。
第二法氫化物發生原子熒光光譜法
9、原理
食品試樣經濕法消解或干灰化法處理后,加亼硫脲使五價砷預還原為三價砷,再加亼硼氫化鈉或硼氫化鉀使還原生成砷化氫,由氬氣載入石英原子化器中分解為原子態砷,在高強度砷空心陰極燈的發射光激發下產生原子熒光,其熒光強度在固定條件下與被測液中的砷濃度成正比,與標準系列比較定量。
10、試劑和材料
注:除非另有說明,本方法所用試劑均為優級純,水為GB/T6682規定的一級水
10.1試劑氫氧化鈉(NaOH)
10.1.2氫氧化鉀(KOH)。
10.1.3硼氫化鉀(KBH4):分析
10.1.4硫脲(CH4N2O2S):分析純。
10.1.5鹽酸(HCl)
10.1.6硝酸(HNO3)
10.1.7硫酸(H2SO4)。
10.1.8高氯酸(HClO4)。
10.1.9硝酸鎂[Mg(NO3)2·6H2O]:分析純。
10.1.10氧化鎂(MgO):分析純
10.1.11抗壞血酸(CH3O3)
10.2試劑配制
10.2.1氫氧化鉀溶液(5g/L):稱取5.0g氫氧化鉀,溶于水并稀釋至1000ml
10.2.2硼氫化鉀溶液(20g/L):稱取硼氫化鉀20.0g,溶于1000mL5g/L氫氧化鉀溶液中,混勻
10.2.3硫脲十抗壞血酸溶液:稱取10.0g硫脲,加約80mL水,加熱溶解,待冷卻后加入10.0g抗壞血酸,稀釋至100mL。現用現配
10.2.4氫氧化鈉溶液(100g/L):稱取10.0g氫氧化鈉,溶于水并稀釋至100mL
10.2.5硝酸鎂溶液(150g/L):稱取15.0g硝酸鎂,溶于水并稀釋至100mL
10.2.6鹽酸溶液(1+1):量取100mL鹽酸,緩緩倒入100mL水中,混勻。
10.2.7硫酸溶液(1+9):量取硫酸100mL,緩緩倒入900mL水中,混勻
10.2.8硝酸溶液(2+98):量取硝酸20mL,緩緩倒入980mL水中,混勻。
10.3標準品
三氧化二砷(As2O3)標準品:純度≥99.5%。
10.4標準溶液配制
10.4.1砷標準儲備液(100mg/L,按As計):準確稱取于100℃干燥2h的三氧化二砷0.0132g,加100g/L氫氧化鈉溶液1mL和少量水溶解,轉入100mL容量瓶中,加人適量鹽酸調整其酸度近中性,加水稀釋至刻度。4℃避光保存,保存期一年。或購買經國家認證并授予標準物質證書的標準溶液物質。
10.4.2砷標準使用液(1.00mg/L,按As計):準確吸取1.00mL砷標準儲備液(100mg/L)于100mL容量瓶中,用硝酸溶液(2+98)稀釋至刻度。現用現配。
11、儀器和設備
注:玻璃器皿及聚四氟乙烯消解內罐均需以硝酸溶液(1+4)浸泡24h,用水反復沖洗,最后用去離子水沖洗干
11.1原子熒光光譜儀
11.2天平:感量為0.1mg和1mg。
11.3組織勻漿器。
11.4高速粉碎機。
11.5控溫電熱板:50℃~200℃。
11.6馬弗爐。
12、分析步驟
12.1試樣預處理見5.1。
12.2試樣消解
12.2.1濕法消解
固體試樣稱取1.0g~2.5g、液體試樣稱取5.0g~10.0g(或mL)(精確至0.001g),置于50mL~100mL錐形瓶中,同時做兩份試劑空白。加硝酸20mL,高氯酸4mL,硫酸1.25mL,放置過夜。次日置于電熱板上加熱消解。若消解液處理至1mL左右時仍有未分解物質或色澤變深,取下放冷,補加硝酸5mL~10mL,再消解至2mL左右,如此反復兩三次,注意避免炭化。繼續加熱至消解完全后,再持續蒸發至高氯酸的白煙散盡,硫酸的白煙開始冒出。冷卻,加水25mL,再蒸發至冒硫酸白煙。冷卻,用水將內溶物轉入25mL容量瓶或比色管中,加入硫脲十抗壞血酸溶液2mL,補加水至刻度,混勻,放置30min,待測。按同一操作方法作空白試驗。
12.2.2干灰化法
固體試樣稱取1.0g~2.5g,液體試樣取4.00ml.(g)(精確至0.001g),置于50mL~100mL坩堝中,同時做兩份試劑空白。加150g/L硝酸鎂10mL混勻,低熱蒸干,將1g氧化鎂覆蓋在干渣上,于電爐上炭化至無黑煙,移入550℃馬弗爐灰化4h。取出放冷,小心加入鹽酸溶液(1+1)10mL以中和氧化鎂并溶解灰分,轉入25mL容量瓶或比色管,向容量瓶或比色管中加入硫脲十抗壞血酸溶液2mL,另用硫酸溶液(1+9)分次洗滌坩堝后合并洗滌液至25mL刻度,混勻,放置30min,待測。按同一操作方法作空白試驗
12.3儀器參考條件
負高壓:260V;砷空心陰極燈電流:50mA~80mA;載氣:氬氣;載氣流速:500mL/min;屏蔽氣流速:800mL/min;測量方式:熒光強度;讀數方式:峰面積。
12.4標準曲線制作
取25mL容量瓶或比色管6支,依次準確加入1.00!g/mL砷標準使用液0.00mL、0.10ml、0.25mL、0.50mL、1.5mL和3.0mL(分別相當于砷濃度0.0ng/mL、4.0ng/mL、10ng/mI、20ng/mL、60ng/mL、120ng/mL),各加硫酸溶液(1+9)12.5mL,硫脲十抗壞血酸溶液2mL,補加水至刻度,混勻后放置30min后測定。
儀器預熱穩定后,將試劑空白、標準系列溶液依次引入儀器進行原子熒光強度的測定。以原子熒光強度為縱坐標,砷濃度為橫坐標繪制標準曲線,得到回歸方程。
12.5試樣溶液的測定
相同條件下,將樣品溶液分別引人儀器進行測定。根據回歸方程計十算出樣品中砷元素的濃度。
13、分析結果的表述

14、精密度
在重復性條件下獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%
15、檢出限
稱樣量為1g,定容體積為25mL時,方法檢出限為0.010mg/kg,方法定量限為0.040mg/kg
第三法銀鹽法
16、原理
試樣經消化后,以碘化鉀、氯化亞錫將高價砷還原為三價砷,然后與鋅粒和酸產生的新生態氫生成砷化氫,經銀鹽溶液吸收后,形成紅色膠態物,與標準系列比較定量
17、試劑和材料
注:除非另有說明,本方法所用試劑均為優級純,水為GB/T6682規定的一級水。
17.1試劑
17.1.1硝酸(HNO3)。
17.1.2硫酸(H2SO4)
17.1.3鹽酸(HCl)
17.1.4高氯酸(HCIO4)。
17.1.5三氯甲烷(CHCl3):分析純。
17.1.6二乙基二硫代氨基甲酸銀[(C2H3)2NCS2Ag]:分析純。
17.1.7氯化亞錫(SnCl2):分析純。
17.1.8硝酸鎂[Mg(NO3)2·6H2O]:分析純。
17.1.9碘化鉀(KI):分析純。
17.1.10氧化鎂(MgO):分析純
17.1.11乙酸鉛(C4H6O4Pb·3H2O):分析純
17.1.12三乙醇胺(C6H15NO3):分析純
17.1.13無砷鋅粒:分析純
17.1.14氫氧化鈉(NaOH)。
17.1.15乙酸。
17.2試劑配制
17.2.1硝酸-高氯酸混合溶液(4+1):量取80mL硝酸,加入20mL高氯酸,混勻。
17.2.2硝酸鎂溶液(150g/L):稱取15g硝酸鎂,加水溶解并稀釋定容至100mL
17.2.3碘化鉀溶液(150g/L):稱取15g碘化鉀,加水溶解并稀釋定容至100mL,貯存于棕色瓶中。
17.2.4酸性氯化亞錫溶液:稱取40g氯化亞錫,加鹽酸溶解并稀釋至100mL,加人數顆金屬錫粒。
17.2.5鹽酸溶液(1+1):量取100mL鹽酸,緩緩倒入100mL水中,混勻。
17.2.6乙酸鉛溶液(100g/L):稱取11.8g乙酸鉛,用水溶解,加入1滴~2滴乙酸,用水稀釋定容至
17.2.7乙酸鉛棉花:用乙酸鉛溶液(100g/L)浸透脫脂棉后,壓除多余溶液,并使之疏松,在100℃C以下干燥后,貯存于玻璃瓶中。
17.28氫氧化鈉溶液(200g/L):稱取20g氫氧化鈉,溶于水并稀釋至100mL。
17.2.9硫酸溶液(6+94):量取6.0mL硫酸,慢慢加入80mL水中,冷卻后再加水稀釋至100mL。
17.2.10二乙基二硫代氨基甲酸銀-三乙醇胺-三氯甲烷溶液:稱取0.25g二乙基二硫代氨基甲酸銀置于乳缽中,加少量三氯甲烷研磨,移入100mL量筒中,加入1.8mL三乙醇胺,再用三氯甲烷分次洗滌乳缽洗滌液一并移入量筒中,用三氯甲烷稀釋至100mL,放置過夜。濾入棕色瓶中貯存。
17.3標準品
三氧化二砷(As2O3)標準品:純度≥99.5%。
17.4標準溶液配制
17.4.1砷標準儲備液(100mg/L,按As計):準確稱取于100℃干燥2h的三氧化二砷0.1320g,加5mL氫氧化鈉溶液(200g/L),溶解后加25mL硫酸溶液(6+94),移入1000mL容量瓶中,加新煮沸冷卻的水稀釋至刻度,貯存于棕色玻塞瓶中。4℃避光保存。保存期一年。或購買經國家認證并授予標準物質證書的標準物質。
17.4.2砷標準使用液(1.00mg/L,按As計):吸取1.00mL砷標準儲備液(100mg/L)于100mL.容量瓶中,加1mL硫酸溶液(6+94),加水稀釋至刻度。
現用現配。
18、儀器和設備
注:所用玻璃器皿均需以硝酸溶液(1+4)浸泡24h,用水反復沖洗,最后用去離子水沖洗干凈
18.1分光光度計。
18.2測砷裝置:見圖
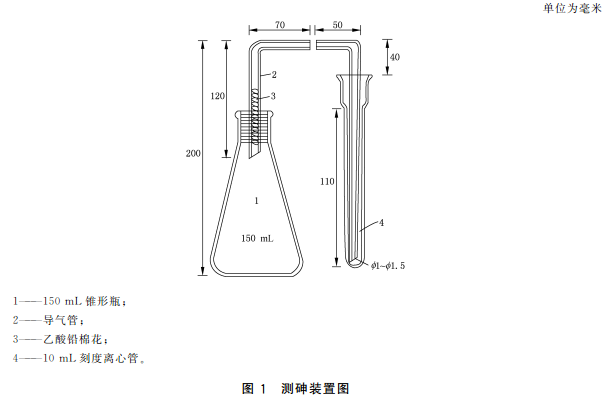
18.2.1 100mL~150mL錐形瓶:19號標準口。
18.2.2導氣管:管口19號標準口或經堿處理后洗凈的橡皮塞與錐形瓶密合時不應漏氣。管的另一端管徑為2.3吸收管:10mL刻度離心管作吸收管用
19、試樣制備
19.1試樣預處理
19.2試樣溶液制備
19.2.1硝酸-高氯酸-硫酸法
19.2.1.1糧食、粉絲、粉條、豆干制品、糕點、茶葉等及其他含水分少的固體食品
稱取5.0g~10.0g試樣(精確至0.001g),置于250mL~500mL定氮瓶中,先加少許水濕潤,加數粒玻璃珠、10mL~15mL硝酸-高氯酸混合液,放置片刻,小火緩緩加熱,待作用緩和,放冷。沿瓶壁加入5mL或10mL硫酸,再加熱,至瓶中液體開始變成棕色時,不斷沿瓶壁滴加硝酸-高氯酸混合液至有機質分解完全。加大火力,至產生白煙,待瓶口白煙冒凈后,瓶內液體再產生白煙為消化完全,該溶液應澄清透明無色或微帶黃色,放冷。(在操作過程中應注意防止爆沸或爆炸)加20mL水煮沸,除去殘余的硝酸至產生白煙為止,如此處理兩次,放冷。將冷后的溶液移入50mL或100mL容量瓶中,用水洗滌定氮瓶,洗滌液并入容量瓶中,放冷,加水至刻度,混勻。定容后的溶液每10mL相當于1g試樣,相當加入硫酸量1mL。取與消化試樣相同量的硝酸-高氯酸混合液和硫酸,按同一方法作空白試驗。
19.2.1.2蔬菜、水果
稱取25.0g~50.0g(精確至0.001g)試樣,置于250mL~500mL定氮瓶中,加數粒玻璃珠、10mL、15mL硝酸-高氯酸混合液,以下按19.2.1.1自“放置片刻”起依法操作,但定容后的溶液每10mL相當于5g試樣,相當于加入硫酸1mL。按同一操作方法作空白試驗
19.2.1.3醬、醬油、醋、冷飲、豆腐、腐乳、醬腌菜等
稱取10.0g~20.0g試樣(精確至0.001g),或吸取10.0mL~20.0mL液體試樣,置于250mL~500mL定氮瓶中,加數粒玻璃珠、5mL~15mL硝酸-高氯酸混合液。以下按19.2.1.1自“放置片刻”起依法操作,但定容后的溶液每10mL相當于2g或2mL試樣。按同一操作方法作空白試驗
19.2.1.4含酒精性飲料或含二氧化碳飲料
吸取10.00mL~20.00mL試樣,置于250mL~500mL定氮瓶中,加數粒玻璃珠,先用小火加熱除去乙醇或二氧化碳,再加5mL~10mL硝酸-高氯酸混合液,混勻后,以下按19.2.1.1自“放置片刻起依法操作,但定容后的溶液每10mL相當于2mL試樣。按同一操作方法作空白試驗。
19.2.1.5含糖量高的食品
稱取5.0g~10.0g試樣(精確至0.001g),置于250mL~500mL定氮瓶中,先加少許水使濕潤,加數粒玻璃珠、5mL~10mL硝酸-高氯酸混合后,搖勻。緩緩加入5mL或10mL硫酸,待作用緩和停止起泡沫后,先用小火緩緩加熱(糖分易炭化),不斷沿瓶壁補加硝酸-高氯酸混合液,待泡沫全部消失后,再加大火力,至有機質分解完全,發生白煙,溶液應澄明無色或微帶黃色,放冷。以下按19.2.1.1自“加20mL水煮沸”起依法操作。按同一操作方法作空白試驗。
19.2.1.6水產品
稱取試樣5.0g~10.0g(精確至0.001g)(海產藻類、貝類可適當減少取樣量),置于250mL~500mL定氮瓶中,加數粒玻璃珠,5mL~10mL硝酸-高氯酸混合液,混勻后,以下按19.2.1.1自“沿瓶壁加入5mL或10mL硫酸”起依法操作。按同一操作方法作空白試驗
19.2.2硝酸-硫酸法
以硝酸代替硝酸-高氯酸混合液進行操作
19.2.3灰化法
19.2.3.1糧食、茶葉及其他含水分少的食品
稱取試樣5.0g(精確至0.001g),置于坩堝中,加1g氧化鎂及10mL硝酸鎂溶液,混勻,浸泡4h。于低溫或置水浴鍋上蒸干,用小火炭化至無煙后移人馬弗爐中加熱至550℃,灼燒3h~4h,冷卻后取出。加5mL水濕潤后,用細玻棒攪拌,再用少量水洗下玻棒上附著的灰分至坩堝內。放水浴上蒸干后移入馬弗爐550℃灰化2h,冷卻后取出。加5mL水濕潤灰分,再慢慢加入10mL鹽酸溶液(1+1),然后將溶液移入50mL容量瓶中,坩堝用鹽酸溶液(1+-1)洗滌3次,每次5mL,再用水洗滌3次,每次5mL,洗滌液均并入容量瓶中,再加水至刻度,混勻。定容后的溶液每10mL相當于1g試樣,其加入鹽酸量不少于(中和需要量除外)1.5mL。全量供銀鹽法測定時,不必再加鹽酸。按同一操作方法作空白試驗。
19.2.3.2植物油
稱取5.0g試樣(精確至0.001g),置于50mL瓷坩堝中,加10g硝酸鎂,再在上面覆蓋2g氧化鎂,將坩堝置小火上加熱,至剛冒煙,立即將坩堝取下,以防內容物溢岀,待煙小后,再加熱至炭化完全將坩堝移至馬弗爐中,550℃以下灼燒至灰化完全,冷后取出。加5mL水濕潤灰分,再緩緩加入15ml鹽酸溶液(1+1),然后將溶液移入50mL容量瓶中,坩堝用鹽酸溶液(1+1)洗滌5次,每次5mL,洗滌液均并入容量瓶中,加鹽酸溶液(1+1)至刻度,混勻。定容后的溶液每10mL相當于1g試樣,相當于加入鹽酸量(中和需要量除外)1.5mL。按同一操作方法作空白試驗。
19.2.3.3水產品
稱取試樣5.0g置于坩堝中(精確至0.001g),加1g氧化鎂及10mL硝酸鎂溶液,混勻,浸泡4h以下按19.2.3.1自“于低溫或置水浴鍋上蒸干”起依法操作。
20、分析步驟
吸取一定量的消化后的定容溶液(相當于5g試樣)及同量的試劑空白液,分別置于150mL錐形瓶中,補加硫酸至總量為5mL,加水至50mL~55mL。
20.1標準曲線的繪制
分別吸取0.0mL、2.0mL、4.0mL、6.0mL、8.0mL、10mL砷標準使用液(相當0.0Pg、2.0"g、4.0g、6.0μg、8.0g、10g)置于6個150mL錐形瓶中,加水至40mL,再加10mL鹽酸溶液(1+1)
20.2用濕法消化液
于試樣消化液、試劑空白液及砷標準溶液中各加3mL碘化鉀溶液(150g/L)、0.5mL酸性氯化亞溶液,混勻,靜置15min。各加入3g鋅粒,立即分別塞上裝有乙酸鉛棉花的導氣管,并使管尖端插入盛有4mL銀鹽溶液的離心管中的液面下,在常溫下反應45min后,取下離心管,加三氯甲烷補足4mL。用1cm比色杯,以零管調節零點,于波長520nm處測吸光度,繪制標準曲線
20.3用灰化法消化液。
取灰化法消化液及試劑空白液分別置于150mL錐形瓶中。吸取0.0mL、2.0mL、4.0mL、6.0mL、8.0mL、10mL砷標準使用液(相當0.0g、2.0Pg、4.0g、6.0g、8.0g、10μg砷),分別置于150mL錐形瓶中,加水至43.5mL,再加6.5mL鹽酸。以下按20.2自“于試樣消化液”起依法操作。
21、分析結果的表述
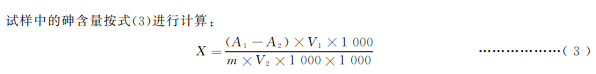

22、精密度
在重復性條件下獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%
23、檢出限
稱樣量為1g,定容體積為25mL時,方法檢出限為0.2mg/kg,方法定量限為0.7mg/kg。
第二篇食品中無機砷的測定
第一法液相色譜-原子熒光光譜法(LC-AFS)法
24、原理
食品中無機砷經稀硝酸提取后,以液相色譜進行分離,分離后的目標化合物在酸性環境下與KBH反應,生成氣態砷化合物,以原子熒光光譜儀進行測定。按保留時間定性,外標法定量。
25、試劑和材料
注:除非另有說明,本方法所用試劑均為優級純,水為GB/T6682規定的一級水
25.1試劑
1.1磷酸二氫銨(NH4H2PO):分析純
25.1.2硼氫化鉀(KBH4):分析純。
25.1.3氫氧化鉀(KOH)
25.1.4硝酸(HNO3)。
25.1.5鹽酸(HCl。
25.1.6氨水(NH3·H2O)
25.1.7正己烷[CH3(CH2)4CH3
25.2試劑配制
25.2.1鹽酸溶液[20%(體積分數)]:量取200mL鹽酸,溶于水并稀釋至1000mL
25.2.2硝酸溶液(0.15mol/L):量取10mL硝酸,溶于水并稀釋至1000mL
25.2.3氫氧化鉀溶液(100g/L):稱取10g氫氧化鉀,溶于水并稀釋至100mlGB5009.11-2014
25.2.4氫氧化鉀溶液(5g/L):稱取5g氫氧化鉀,溶于水并稀釋至1000mL
25.2.5硼氫化鉀溶液(30g/L):稱取30g硼氫化鉀,用5g/L氫氧化鉀溶液溶解并定容至1000mL,現用現配。
25.2.6磷酸二氫銨溶液(20mmol/L):稱取2.3g磷酸二氫銨,溶于1000mL水中,以氨水調節pH至8.0,經0.45m水系濾膜過濾后,于超聲水浴中超聲脫氣30min,備用
25.2.7磷酸二氫銨溶液(1mmol/L):量取20mmol/L磷酸二氫銨溶液50mL,水稀釋至1000mL,以氨水調pH至9.0,經0.45m水系濾膜過濾后,于超聲水浴中超聲脫氣30min,備用
25.2.8磷酸二氫銨溶液(15mmol/L):稱取1.7g磷酸二氫銨,溶于1000mL水中,以氨水調節pH至6.0,經0.45m水系濾膜過濾后,于超聲水浴中超聲脫氣30min,備用
25.3標準品
25.3.1氧化二砷(AS2O3)標準品:純度≥99.5%。
25.3.2砷酸二氫鉀(KH2AsO4)標準品:純度≥99.5%。
25.4標準溶液配制
25.4.1亞砷酸鹽[As(Ⅲ)標準儲備液(100mg/L,按As計):準確稱取三氧化二砷0.0132g,加100g/L氫氧化鉀溶液1mL和少量水溶解,轉入100mL容量瓶中,加入適量鹽酸調整其酸度近中性,加水稀釋至刻度。4℃保存,保存期一年。或購買經國家認證并授予標準物質證書的標準溶液物質
25.4.2砷酸鹽[As(V)]標準儲備液(100mg/L,按As計):準確稱取砷酸二氫鉀0.0240g,水溶解,轉入100mL容量瓶中并用水稀釋至刻度。4℃保存,保存期一年。或購買經國家認證并授予標準物質證書的標準溶液物質。
25.4.3As(Ⅲ)、As(V)混合標準使用液(1.00mg/L,按As計):分別準確吸取1.0mLAs(Ⅲ)標準儲備液(100mg/L)、1.0mLAs(V)標準儲備液(100mg/L)于100mL容量瓶中,加水稀釋并定容至刻度。現用現配。
26、儀器和設備
注:所用玻璃器皿均需以硝酸溶液(1+4)浸泡24h,用水反復沖洗,最后用去離子水沖洗干凈
26.1液相色譜原子熒光光譜聯用儀(LC-AFS):由液相色譜儀(包括液相色譜泵和手動進樣閥)與原子熒光光譜儀組成。
26.2組織勻漿器。
26.3高速粉碎機。
26.4冷凍干燥機
26.5離心機:轉速≥8000r/min
26.6pH計:精度為0.01。
26.7天平:感量為0.1mg和1mg
26.8恒溫干燥箱(50℃~300℃℃)。
26.9C18凈化小柱或等效柱
27、分析步驟
27.1試樣預處理見5.1。
27.2試樣提取
27.2.1稻米樣品
稱取約1.0g稻米試樣(準確至0.001g)于50mL塑料離心管中,加入20mL0.15mol/L硝酸溶液,放置過夜。于90℃恒溫箱中熱浸提2.5h,每0.5h振搖1min。提取完畢,取出冷卻至室溫,8000r/min離心15min,取上層清液,經0.45m有機濾膜過濾后進樣測定。按同一操作方法作空白試驗。
27.2.2水產動物樣品
稱取約1.0g水產動物濕樣(準確至0.001g),置于50mL塑料離心管中,加入20mL0.15mol/L硝酸溶液,放置過夜。于90℃恒溫箱中熱浸提2.5h,每0.5h振搖1min。提取完畢,取出冷卻至室溫,8000r/min離心15min。取5mL上清液置于離心管中,加入5mL正己烷,振搖1min后,8000r/min離心15min,棄去上層正己烷。按此過程重復一次。吸取下層清液,經0.45!m有機濾膜過濾及C18小柱凈化后進樣。按同一操作方法作空白試驗
27.2.3嬰幼兒輔助食品樣品
稱取嬰幼兒輔助食品約1.0g(準確至0.001g)于15mL塑料離心管中,加入10mL0.15mol/L硝酸溶液,放置過夜。于90℃恒溫箱中熱浸提2.5h,每0.5h振搖1min,提取完畢,取出冷卻至室溫。8000r/min離心15min。取5mL上清液置于離心管中,加入5mL正己烷,振搖1min,8000r/min離心15min,棄去上層正己烷。按此過程重復一次。吸取下層清液,經0.45m有機濾膜過濾及C18小柱凈化后進行分析。按同一操作方法作空白試驗
27.3儀器參考條件
27.3.1液相色譜參考條件
色譜柱:陰離子交換色譜柱(柱長250mm,內徑4mm),或等效柱。陰離子交換色譜保護柱(柱長10mm,內徑4mm),或等效柱流動相組成a)等度洗脫流動相:15mmol/L磷酸二氫銨溶液(pH6.0),流動相洗脫方式等度洗脫。流動相流速:1.0mL/min;進樣體積:100!L。等度洗脫適用于稻米及稻米加工食品b)梯度洗脫:流動相A:1mmol/L磷酸二氫銨溶液(pH9.0);流動相B:20mmol/L磷酸二氫銨溶液(pH8.0)。(梯度洗脫程序見附錄A中的表A.4。)流動相流速:1.0mL/min;進樣體積100μL。梯度洗脫適用于水產動物樣品、含水產動物組成的樣品、含藻類等海產植物的樣品以及嬰。
27.3.2原子熒光檢測參考條件
負高壓:320V;砷燈總電流:90mA;主電流/輔助電流:55/35;原子化方式:火焰原子化;原子化器溫度:中溫。
載液:20%鹽酸溶液,流速4mL/min;還原劑:30g/L硼氫化鉀溶液,流速4mL/min;載氣流速400mL/min;輔助氣流速:400mL/min
27.4標準曲線制作
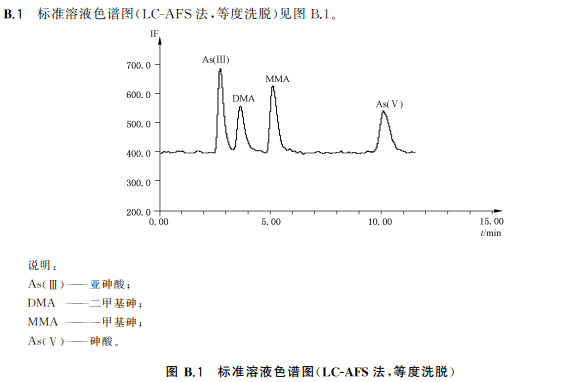
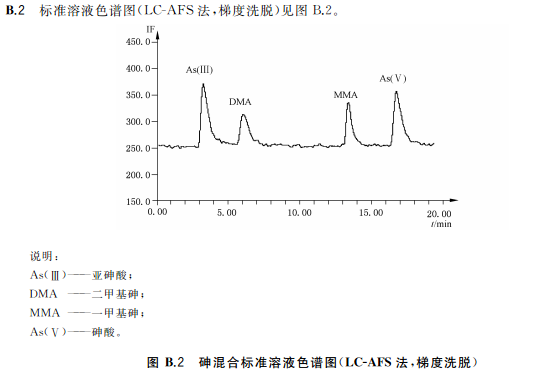
取7支10mL容量瓶,分別準確加入1.00mg/L混合標準使用液0.00mL、0.050mL、O.10mL、0.20mL、0.30mL、0.50mL和1.0mL,加水稀釋至刻度,此標準系列溶液的濃度分別為0.0ng/mL、5.0ng/mL、10ng/mL、20ng/mL、30ng/mL、50ng/mL和100ng/mL吸取標準系列溶液100μL注入液相色譜-原子熒光光譜聯用儀進行分析,得到色譜圖,以保留時間定性。以標準系列溶液中目標化合物的濃度為橫坐標,色譜峰面積為縱坐標,繪制標準曲線。標準溶液色譜圖見附錄B中的圖B.1、圖B.2。
27.5試樣溶液的測定
吸取試樣溶液100L注入液相色譜-原子熒光光譜聯用儀中,得到色譜圖,以保留時間定性。根據標準曲線得到試樣溶液中As(Ⅲ)與As(V)含量,As(Ⅲ)與As(V)含量的加和為總無機砷含量,平行測定次數不少于兩次。
28、分析結果的表述

29、精密度
在重復性條件下獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%。
30、其他
本方法檢出限:取樣量為1g,定容體積為20mL時,檢出限為:稻米0.02mg/kg、水產動物03mg/kg、嬰幼兒輔助食品0.02mg/kg;定量限為:稻米0.05mg/kg、水產動物0.08mg/kg、嬰幼兒輔助食品0.05mg/kg
第二法液相色譜-電感耦合等離子質譜法(LC-ICP/MS)
31、原理
食品中無機砷經稀硝酸提取后,以液相色譜進行分離,分離后的目標化合物經過霧化由載氣送入ICP炬焰中,經過蒸發、解離、原子化、電離等過程,大部分轉化為帶正電荷的正離子,經離子采集系統進人質譜儀,質譜儀根據質荷比進行分離測定。以保留時間定性和質荷比定性,外標法定量。
32、試劑和材料
注:除非另有說明,本方法所用試劑均為優級純,水為GB/T6682規定的一級水
32.1試劑
32.1.1無水乙酸鈉(NaCH3COO):分析純
32.1.2硝酸鉀(KNO3):分析純
32.1.3磷酸二氫鈉(NaH2PO4):分析純
32.1.4乙二胺四乙酸二鈉(C10H14N2Na2O):分析純。
32.1.5硝酸(HNO3)。
32.1.6正己烷[CH3(CH2)4CH3]。
32.1.7無水乙醇(CH3CH2OH)
32.1.8氨水(NH3·H2O)
32.2試劑配制
32.2.1硝酸溶液(0.15mol/L):量取10mL硝酸,加水稀釋至1000mL
322.2流動相A相:含10mmol/L無水乙酸鈉、3mmol/L硝酸鉀、10mmol/L磷酸二氫鈉、0.2mmol/L乙二胺四乙酸二鈉的緩沖液(pH10)。分別準確稱取0.820g無水乙酸鈉、0.303g硝酸鉀、1.56g磷酸二氫鈉、0.075g乙二胺四乙酸二鈉,用水定容值1000mL,氨水調節pH為10,混勻。經45m水系濾膜過濾后,于超聲水浴中超聲脫氣30min,備用32.2.3氫氧化鉀溶液(100g/L):稱取10g氫氧化鉀,加水溶解并稀釋至100mL
32.3標準品
32.3.1氧化二砷(As2O3)標準品:純度≥99.5%
32.3.2砷酸二氫鉀(KH2AsO4)標準品:純度≥99.5%
32.4標準溶液配制
32.4.1亞砷酸鹽[As(Ⅲ)]標準儲備液(100mg/L,按As計):準確稱取三氧化二砷0.0132g,加1mL氫氧化鉀溶液(100g/L)和少量水溶解,轉入100mL容量瓶中,加入適量鹽酸調整其酸度近中性,加水稀釋至刻度。4℃保存,保存期一年。或購買經國家認證并授予標準物質證書的標準溶液物質
32.4.2砷酸鹽[As(Ⅴ)標準儲備液(100mg/L,按As計):準確稱取砷酸二氫鉀0.0240g,水溶解,轉入100mL容量瓶中并用水稀釋至刻度。4℃保存,保存期一年。或購買經國家認證并授予標準物質證書的標準物質。
32.4.3As(Ⅲ)、As(V)混合標準使用液(1.00mg/L,按As計):分別準確吸取1.0mLAs(Ⅲ)標準儲備液(100mg/L)、1.0mLAs(V)標準儲備液(100mg/L)于100mL容量瓶中,加水稀釋并定容至刻度。現用現配。
33、儀器和設備
注:所用玻璃器皿均需以硝酸溶液(1+4)浸泡24h,用水反復沖洗,最后用去離子水沖洗干凈
33.1液相色譜-電感耦合等離子質譜聯用儀(IC-ICP/MS):由液相色譜儀與電感耦合等離子質譜儀組成
33.2組織勻漿器
33.3高速粉碎機。
33.4冷凍干燥機。
33.5離心機:轉速≥8000r/min
33.6pH計:精度為0.01
33.7天平:感量為0.1mg和1mg
33.8恒溫干燥箱(50℃~300℃C)
34、分析步驟
34.1試樣預處理
34.2試樣提取
34.2.1稻米樣品見27.2.1
34.2.2水產動物樣見27.2.2。
34.2.3嬰幼兒輔助食品樣品見27.2.3。
34.3儀器參考條件
34.3.1液相色譜參考條件
色譜柱:陰離子交換色譜分析柱(柱長250mm,內徑4mm),或等效柱。陰離子交換色譜保護柱柱長10mm,內徑4mm)或等效柱。流動相:(含10mmol/L無水乙酸鈉、3mmol/L硝酸鉀、10mmol/L磷酸二氫鈉、0.2mmol/L乙二胺四乙酸二鈉的緩沖液,氨水調節pH為10):無水乙醇=99:1(體積比)。洗脫方式:等度洗脫。進樣體積:50L
34.3.2電感耦合等離子體質譜儀參考條件
RF入射功率1550W;載氣為高純氬氣;載氣流速0.85L/min;補償氣0.15L/min。泵速0.3rps檢測質量數m/z=75(As),m/z=35(Cl)。
34.4標準曲線制作
分別準確吸取1.00mg/L混合標準使用液0.00mL、0.025mL、0.050mL、0.10mL、0.50mL和1.0mL于6個10mL容量瓶,用水稀釋至刻度,此標準系列溶液的濃度分別為0.0ng/mL、2.5ng/mL5ng/mL、10ng/mL、50ng/mL和100ng/mL。
用調諧液調整儀器各項指標,使儀器靈敏度、氧化物、雙電荷、分辨率等各項指標達到測定要求。
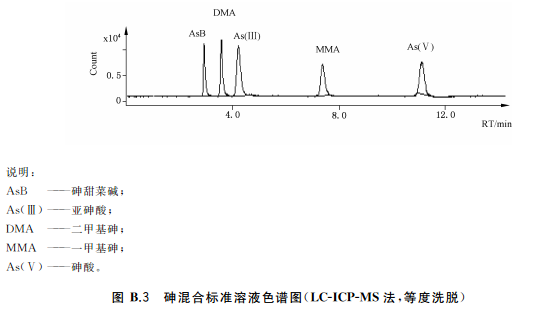
吸取標準系列溶液50μL注入液相色譜-電感耦合等離子質譜聯用儀,得到色譜圖,以保留時間定性。以標準系列溶液中目標化合物的濃度為橫坐標,色譜峰面積為縱坐標,繪制標準曲線。標準溶液色譜圖見附錄B中的圖B.3
34.5試樣溶液的測定
吸取試樣溶液50L注入液相色譜-電感耦合等離子質譜聯用儀,得到色譜圖,以保留時間定性。根據標準曲線得到試樣溶液中As(Ⅲ)與As(V)含量,As(Ⅲ)與As(V)含量的加和為總無機砷含量平行測定次數不少于兩次。
35、分析結果的表述

36、精密度
在重復性條件獲得的兩次獨立測定結果的絕對差值不得超過算術平均值的20%
37、其他
本方法檢出限:取樣量為1g,定容體積為20mL時,方法檢出限為:稻米0.01mg/kg、水產動物0.02mg/kg、嬰幼兒輔助食品0.01mg/kg;方法定量限為:稻米0.03mg/kg、水產動物0.06mg/kg、嬰幼兒輔助食品0.03mg/kg
附錄A
微波消解參考條件
附錄B
色譜圖
上一篇:無

掃描二維碼
版權所有©廣州格丹納儀器有限公司 ![]() 粵公網安備 44011202000628號 粵ICP備14047970號
粵公網安備 44011202000628號 粵ICP備14047970號
分享到:







