用領(lǐng)域")

地址:廣州市南沙區(qū)大崗鎮(zhèn)智新一路8號(hào)聯(lián)東U谷廣州南沙國(guó)際企業(yè)港7棟5樓
產(chǎn)品咨詢(xún):13392693259
耗材電話:18998328368
售后服務(wù):020-87684117
傳 真:020-87684846
Email:info@gdana.com
網(wǎng)址:http://www.yytqd.com.cn/
前言
本標(biāo)準(zhǔn)代替GB5009.12—2010《食品安全國(guó)家標(biāo)準(zhǔn) 食品中鉛的測(cè)定》、GB/T20380.3—2006《淀粉及其制品 重金屬含量 第3部分:電熱原子吸收光譜法測(cè)定鉛含量》、GB/T23870—2009《蜂膠中鉛的測(cè)定 微波消解-石墨爐原 子吸收分光光度法》、GB/T18932.12—2002《蜂蜜中鉀、鈉、鈣、鎂、鋅、鐵、銅、錳、鉻、鉛、鎘含量的測(cè)定方法 原子吸收光譜法》、NY/T1100—2006《稻米中鉛、鎘的測(cè)定 石墨爐原子吸收光譜法》,SN/T2211—2008《蜂皇漿中鉛和鎘的測(cè)定石墨爐原子吸收光譜法》中鉛的測(cè)定方法。
本標(biāo)準(zhǔn)與GB5009.12—2010相比,主要變化如下:
——在前處理方法中,保留濕法消解和壓力罐消解,刪除干法灰化和過(guò)硫酸銨灰化法,增加微波消解;
——保留石墨爐原子吸收光譜法為第一法,采用磷酸二氫銨-硝酸鈀溶液作為基體改進(jìn)劑;保留火焰原子吸收光 譜法為第三法;保留二硫腙比色法為第四法;
——增加電感耦合等離子體質(zhì)譜法作為第二法; ——?jiǎng)h除氫化物原子熒光光譜法、單掃描極譜法;
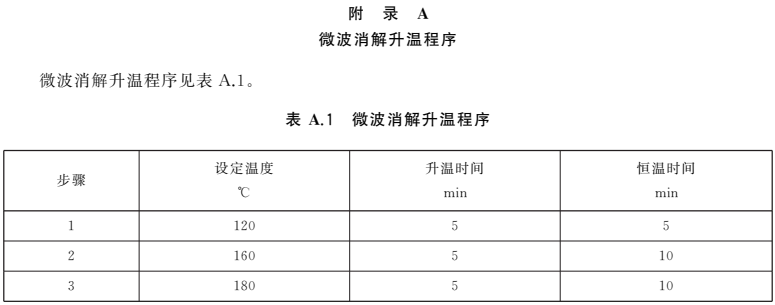
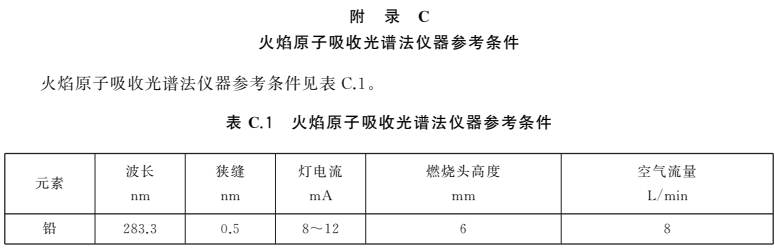
——增加了微波消解升溫程序、石墨爐原子吸收光譜法和火焰原子吸收光譜法的儀器參考條件為附錄。
1、范圍
本標(biāo)準(zhǔn)規(guī)定了食品中鉛含量測(cè)定的石墨爐原子吸收光譜法、電感耦合等離子體質(zhì)譜法、火焰原子吸收光譜法和二硫腙比色法。
本標(biāo)準(zhǔn)適用于各類(lèi)食品中鉛含量的測(cè)定。
第一法石墨爐原子吸收光譜法
2、原理
試樣消解處理后,經(jīng)石墨爐原子化,在283.3nm處測(cè)定吸光度。在一定濃度范圍內(nèi)鉛的吸光度值與鉛含量成正比,與標(biāo)準(zhǔn)系列比較定量。
3、試劑和材料
除非另有說(shuō)明,本方法所用試劑均為優(yōu)級(jí)純,水為GB/T6682規(guī)定的二級(jí)水。
3.1試劑
3.1.1硝酸(HNO3)。
3.1.2高氯酸(HClO4)。
3.1.3磷酸二氫銨(NH4H2PO4)。
3.1.4硝酸鈀[Pd(NO3)2]。
3.2試劑配制
3.2.1硝酸溶液(5+95):量取50mL硝酸,緩慢加入到950mL水中,混勻。
3.2.2硝酸溶液(1+9):量取50mL硝酸,緩慢加入到450mL水中,混勻。
3.2.3磷酸二氫銨-硝酸鈀溶液:稱(chēng)取0.02g硝酸鈀,加少量硝酸溶液(1+9)溶解后,再加入2g磷酸二氫銨,溶解后用硝酸溶液(5+95)定容至100mL,混勻。
3.3標(biāo)準(zhǔn)品 硝酸鉛[Pb(NO3)2,CAS號(hào):10099-74-8]:純度>99.99%。或經(jīng)國(guó)家認(rèn)證并授予標(biāo)準(zhǔn)物質(zhì)證書(shū)的一定濃度的鉛標(biāo)準(zhǔn)溶液。
3.4標(biāo)準(zhǔn)溶液配制
3.4.1鉛標(biāo)準(zhǔn)儲(chǔ)備液(1000mg/L):準(zhǔn)確稱(chēng)取1.5985g(精確至0.0001g)硝酸鉛,用少量硝酸溶液(1+9)溶解,移入1000mL容量瓶,加水至刻度,混勻。 3.4.2鉛標(biāo)準(zhǔn)中間液(1.00mg/L):準(zhǔn)確吸取鉛標(biāo)準(zhǔn)儲(chǔ)備液(1000mg/L)1.00mL于1000mL容量瓶中,加硝酸溶液(5+95)至刻度,混勻。
3.4.3鉛標(biāo)準(zhǔn)系列溶液:分別吸取鉛標(biāo)準(zhǔn)中間液(1.00mg/L)0mL、0.500mL、1.00mL、2.00mL、3.00mL和4.00mL于100mL容量瓶中,加硝酸溶液(5+95)至刻度,混勻。此鉛標(biāo)準(zhǔn)系列溶液的質(zhì)量濃度分別為0μg/L、5.00μg/L、10.0μg/L、20.0μg/L、30.0μg/L和40.0μg/L。
注:可根據(jù)儀器的靈敏度及樣品中鉛的實(shí)際含量確定標(biāo)準(zhǔn)系列溶液中鉛的質(zhì)量濃度。
4、儀器和設(shè)備
注:所有玻璃器皿及聚四氟乙烯消解內(nèi)罐均需硝酸溶液(1+5)浸泡過(guò)夜,用自來(lái)水反復(fù)沖洗,最后用水沖洗干凈。
4.1原子吸收光譜儀:配石墨爐原子化器,附鉛空心陰極燈。
4.2分析天平:感量0.1mg和1mg。
4.3可調(diào)式電熱爐。
4.4可調(diào)式電熱板。
4.5微波消解系統(tǒng):配聚四氟乙烯消解內(nèi)罐。
4.6恒溫干燥箱。
4.7壓力消解罐:配聚四氟乙烯消解內(nèi)罐。
5、分析步驟
5.1試樣制備
注:在采樣和試樣制備過(guò)程中,應(yīng)避免試樣污染。
5.1.1糧食、豆類(lèi)樣品
樣品去除雜物后,粉碎,儲(chǔ)于塑料瓶中。
5.1.2蔬菜、水果、魚(yú)類(lèi)、肉類(lèi)等樣品
樣品用水洗凈,晾干,取可食部分,制成勻漿,儲(chǔ)于塑料瓶中。
5.1.3飲料、酒、醋、醬油、食用植物油、液態(tài)乳等液體樣品
將樣品搖勻。
5.2試樣前處理
5.2.1濕法消解
稱(chēng)取固體試樣0.2g~3g(精確至0.001g)或準(zhǔn)確移取液體試樣0.500mL~5.00mL于帶刻度消化管中,加入10mL硝酸和0.5mL高氯酸,在可調(diào)式電熱爐上消解(參考條件:120℃/0.5h~1h;升至180℃/2h~4h、升至200℃~220℃)。若消化液呈棕褐色,再加少量硝酸,消解至冒白煙,消化液呈無(wú)色透明或略帶黃色,取出消化管,冷卻后用水定容至10mL,混勻備用。同時(shí)做試劑空白試驗(yàn)。亦可采用錐形瓶,于可調(diào)式電熱板上,按上述操作方法進(jìn)行濕法消解。
5.2.2微波消解
稱(chēng)取固體試樣0.2g~0.8g(精確至0.001g)或準(zhǔn)確移取液體試樣0.500mL~3.00mL于微波消解罐中,加入5mL硝酸,按照微波消解的操作步驟消解試樣,消解條件參考附錄A。冷卻后取出消解罐,在電熱板上于140℃~160℃趕酸至1mL左右。消解罐放冷后,將消化液轉(zhuǎn)移至10mL容量瓶中,用少量水洗滌消解罐2次~3次,合并洗滌液于容量瓶中并用水定容至刻度,混勻備用。同時(shí)做試劑空白試驗(yàn)。
5.2.3壓力罐消解
稱(chēng)取固體試樣0.2g~1g(精確至0.001g)或準(zhǔn)確移取液體試樣0.500mL~5.00mL于消解內(nèi)罐中,加入5mL硝酸。蓋好內(nèi)蓋,旋緊不銹鋼外套,放入恒溫干燥箱,于140℃~160℃下保持4h~5h。冷卻后緩慢旋松外罐,取出消解內(nèi)罐,放在可調(diào)式電熱板上于140℃~160℃趕酸至1mL左右。冷卻后將消化液轉(zhuǎn)移至10mL容量瓶中,用少量水洗滌內(nèi)罐和內(nèi)蓋2次~3次,合并洗滌液于容量瓶中并用水定容至刻度,混勻備用。同時(shí)做試劑空白試驗(yàn)。
5.3測(cè)定
5.3.1儀器參考條件
根據(jù)各自?xún)x器性能調(diào)至最佳狀態(tài)。參考條件見(jiàn)附錄B。
5.3.2標(biāo)準(zhǔn)曲線的制作
按質(zhì)量濃度由低到高的順序分別將10μL鉛標(biāo)準(zhǔn)系列溶液和5μL磷酸二氫銨-硝酸鈀溶液(可根據(jù)所使用的儀器確定最佳進(jìn)樣量)同時(shí)注入石墨爐,原子化后測(cè)其吸光度值,以質(zhì)量濃度為橫坐標(biāo),吸光度值為縱坐標(biāo),制作標(biāo)準(zhǔn)曲線。
5.3.3試樣溶液的測(cè)定
在與測(cè)定標(biāo)準(zhǔn)溶液相同的實(shí)驗(yàn)條件下,將10μL空白溶液或試樣溶液與5μL磷酸二氫銨-硝酸鈀溶液(可根據(jù)所使用的儀器確定最佳進(jìn)樣量)同時(shí)注入石墨爐,原子化后測(cè)其吸光度值,與標(biāo)準(zhǔn)系列比較定量。
6、分析結(jié)果的表述
試樣中鉛的含量按式(1)計(jì)算:
X=(ρ-ρ0)×V/m×1000…………………………(1)
式中:
X——試樣中鉛的含量,單位為毫克每千克或毫克每升(mg/kg或mg/L);
ρ——試樣溶液中鉛的質(zhì)量濃度,單位為微克每升(μg/L);
ρ0——空白溶液中鉛的質(zhì)量濃度,單位為微克每升(μg/L);
V——試樣消化液的定容體積,單位為毫升(mL);
m——試樣稱(chēng)樣量或移取體積,單位為克或毫升(g或mL);
1000——換算系數(shù)。
當(dāng)鉛含量≥1.00mg/kg(或mg/L)時(shí),計(jì)算結(jié)果保留三位有效數(shù)字;當(dāng)鉛含量<1.00mg/kg(或mg/L)時(shí),計(jì)算結(jié)果保留兩位有效數(shù)字。
7、精密度
在重復(fù)性條件下獲得的兩次獨(dú)立測(cè)定結(jié)果的絕對(duì)差值不得超過(guò)算術(shù)平均值的20%。
8、其他
當(dāng)稱(chēng)樣量為0.5g(或0.5mL),定容體積為10mL時(shí),方法的檢出限為0.02mg/kg(或0.02mg/L),定量限為0.04mg/kg(或0.04mg/L)。
第二法電感耦合等離子體質(zhì)譜法
見(jiàn)GB5009.268。
第三法火焰原子吸收光譜法
9、原理
試樣經(jīng)處理后,鉛離子在一定pH條件下與二乙基二硫代氨基甲酸鈉(DDTC)形成絡(luò)合物,經(jīng)4-甲基-2-戊酮(MIBK)萃取分離,導(dǎo)入原子吸收光譜儀中,經(jīng)火焰原子化,在283.3nm處測(cè)定的吸光度。在一定濃度范圍內(nèi)鉛的吸光度值與鉛含量成正比,與標(biāo)準(zhǔn)系列比較定量。
10、試劑和材料
注:除非另有說(shuō)明,本方法所用試劑均為分析純,水為GB/T6682規(guī)定的二級(jí)水。
10.1試劑
10.1.1硝酸(HNO3):優(yōu)級(jí)純。
10.1.2高氯酸(HClO4):優(yōu)級(jí)純。
10.1.3硫酸銨[(NH4)2SO4]。
10.1.4檸檬酸銨[C6H5O7(NH4)3]。
10.1.5溴百里酚藍(lán)(C27H28O5SBr2)。
10.1.6二乙基二硫代氨基甲酸鈉[DDTC,(C2H5)2NCSSNa·3H2O]。
10.1.7氨水(NH3·H2O):優(yōu)級(jí)純。
10.1.84-甲基-2-戊酮(MIBK,C6H12O)。
10.1.9鹽酸(HCl):優(yōu)級(jí)純。
10.2試劑配制
10.2.1硝酸溶液(5+95):量取50mL硝酸,加入到950mL水中,混勻。
10.2.2硝酸溶液(1+9):量取50mL硝酸,加入到450mL水中,混勻。
10.2.3硫酸銨溶液(300g/L):稱(chēng)取30g硫酸銨,用水溶解并稀釋至100mL,混勻。
10.2.4檸檬酸銨溶液(250g/L):稱(chēng)取25g檸檬酸銨,用水溶解并稀釋至100mL,混勻。
10.2.5溴百里酚藍(lán)水溶液(1g/L):稱(chēng)取0.1g溴百里酚藍(lán),用水溶解并稀釋至100mL,混勻。
10.2.6DDTC溶液(50g/L):稱(chēng)取5gDDTC,用水溶解并稀釋至100mL,混勻。 10.2.7氨水溶液(1+1):吸取100mL氨水,加入100mL水,混勻。
10.2.8鹽酸溶液(1+11):吸取10mL鹽酸,加入110mL水,混勻。
10.3標(biāo)準(zhǔn)品
硝酸鉛[Pb(NO3)2,CAS號(hào):10099-74-8]:純度>99.99%。或經(jīng)國(guó)家認(rèn)證并授予標(biāo)準(zhǔn)物質(zhì)證書(shū)的一定濃度的鉛標(biāo)準(zhǔn)溶液。
10.4標(biāo)準(zhǔn)溶液配制
10.4.1鉛標(biāo)準(zhǔn)儲(chǔ)備液(1000mg/L):準(zhǔn)確稱(chēng)取1.5985g(精確至0.0001g)硝酸鉛,用少量硝酸溶液(1+9)溶解,移入1000mL容量瓶,加水至刻度,混勻。
10.4.2鉛標(biāo)準(zhǔn)使用液(10.0mg/L):準(zhǔn)確吸取鉛標(biāo)準(zhǔn)儲(chǔ)備液(1000mg/L)1.00mL于100mL容量瓶中,加硝酸溶液(5+95)至刻度,混勻。
11、儀器和設(shè)備
注:所有玻璃器皿均需硝酸(1+5)浸泡過(guò)夜,用自來(lái)水反復(fù)沖洗,最后用水沖洗干凈。
11.1原子吸收光譜儀:配火焰原子化器,附鉛空心陰極燈。
11.2分析天平:感量0.1mg和1mg。
11.3可調(diào)式電熱爐。
11.4可調(diào)式電熱板。
12、分析步驟
12.1試樣制備
同5.1。
12.2試樣前處理
同5.2.1
12.3測(cè)定
12.3.1儀器參考條件
根據(jù)各自?xún)x器性能調(diào)至最佳狀態(tài)。參考條件參見(jiàn)附錄C。
12.3.2標(biāo)準(zhǔn)曲線的制作
分別吸取鉛標(biāo)準(zhǔn)使用液0mL、0.250mL、0.500mL、1.00mL、1.50mL和2.00mL(相當(dāng)0μg、2.50μg、5.00μg、10.0μg、15.0μg和20.0μg鉛)于125mL分液漏斗中,補(bǔ)加水至60mL。加2mL檸檬酸銨溶液(250g/L),溴百里酚藍(lán)水溶液(1g/L)3滴~5滴,用氨水溶液(1+1)調(diào)pH至溶液由黃變藍(lán),加硫酸銨溶液(300g/L)10mL,DDTC溶液(1g/L)10mL,搖勻。放置5min左右,加入10mLMIBK,劇烈振搖提取1min,靜置分層后,棄去水層,將MIBK層放入10mL帶塞刻度管中,得到標(biāo)準(zhǔn)系列溶液。
將標(biāo)準(zhǔn)系列溶液按質(zhì)量由低到高的順序分別導(dǎo)入火焰原子化器,原子化后測(cè)其吸光度值,以鉛的質(zhì)量為橫坐標(biāo),吸光度值為縱坐標(biāo),制作標(biāo)準(zhǔn)曲線。
12.3.3試樣溶液的測(cè)定
將試樣消化液及試劑空白溶液分別置于125mL分液漏斗中,補(bǔ)加水至60mL。加2mL檸檬酸銨溶液(250g/L),溴百里酚藍(lán)水溶液(1g/L)3滴~5滴,用氨水溶液(1+1)調(diào)pH至溶液由黃變藍(lán),加硫酸銨溶液(300g/L)10mL,DDTC溶液(1g/L)10mL,搖勻。放置5min左右,加入10mLMIBK,劇烈振搖提取1min,靜置分層后,棄去水層,將MIBK層放入10mL帶塞刻度管中,得到試樣溶液和空白溶液。
將試樣溶液和空白溶液分別導(dǎo)入火焰原子化器,原子化后測(cè)其吸光度值,與標(biāo)準(zhǔn)系列比較定量。
13、分析結(jié)果的表述
試樣中鉛的含量按式(2)計(jì)算:
X=(m1-m0)/m2…………………………(2)
式中:
X———試樣中鉛的含量,單位為毫克每千克或毫克每升(mg/kg或mg/L);
m1———試樣溶液中鉛的質(zhì)量,單位為微克(μg);
m0———空白溶液中鉛的質(zhì)量,單位為微克(μg);
m2———試樣稱(chēng)樣量或移取體積,單位為克或毫升(g或mL)。
當(dāng)鉛含量≥10.0mg/kg(或mg/L)時(shí),計(jì)算結(jié)果保留三位有效數(shù)字;當(dāng)鉛含量<10.0mg/kg(或mg/L)時(shí),計(jì)算結(jié)果保留兩位有效數(shù)字。
14、精密度
在重復(fù)性條件下獲得的兩次獨(dú)立測(cè)定結(jié)果的絕對(duì)差值不得超過(guò)算術(shù)平均值的20%。
15、其他
以稱(chēng)樣量0.5g(或0.5mL)計(jì)算,方法的檢出限為0.4mg/kg(或0.4mg/L),定量限為1.2mg/kg(或1.2mg/L)。
第四法二硫腙比色法
16、原理
試樣經(jīng)消化后,在pH8.5~9.0時(shí),鉛離子與二硫腙生成紅色絡(luò)合物,溶于三氯甲烷。加入檸檬酸銨、氰化鉀和鹽酸羥胺等,防止鐵、銅、鋅等離子干擾。于波長(zhǎng)510nm處測(cè)定吸光度,與標(biāo)準(zhǔn)系列比較定量。
17、試劑和材料
除非另有說(shuō)明,本方法所用試劑均為分析純,水為GB/T6682規(guī)定的三級(jí)水。
17.1試劑
17.1.1硝酸(HNO3):優(yōu)級(jí)純。
17.1.2高氯酸(HClO4):優(yōu)級(jí)純。
17.1.3氨水(NH3·H2O):優(yōu)級(jí)純。
17.1.4鹽酸(HCl):優(yōu)級(jí)純。
17.1.5酚紅(C19H14O5S)。
17.1.6鹽酸羥胺(NH2OH·HCl)。
17.1.7檸檬酸銨[C6H5O7(NH4)3]。
17.1.8氰化鉀(KCN)。
17.1.9三氯甲烷(CH3Cl,不應(yīng)含氧化物)。
17.1.10二硫腙(C6H5NHNHCSN=NC6H5)。
17.1.11乙醇(C2H5OH):優(yōu)級(jí)純。
17.2試劑配制
17.2.1硝酸溶液(5+95):量取50mL硝酸,緩慢加入到950mL水中,混勻。
17.2.2硝酸溶液(1+9):量取50mL硝酸,緩慢加入到450mL水中,混勻。
17.2.3氨水溶液(1+1):量取100mL氨水,加入100mL水,混勻。
17.2.4氨水溶液(1+99):量取10mL氨水,加入990mL水,混勻。
17.2.5鹽酸溶液(1+1):量取100mL鹽酸,加入100mL水,混勻。
17.2.6酚紅指示液(1g/L):稱(chēng)取0.1g酚紅,用少量多次乙醇溶解后移入100mL容量瓶中并定容至刻度,混勻。
17.2.7二硫腙-三氯甲烷溶液(0.5g/L):稱(chēng)取0.5g二硫腙,用三氯甲烷溶解,并定容至1000mL,混勻,保存于0℃~5℃下,必要時(shí)用下述方法純化。 稱(chēng)取0.5g研細(xì)的二硫腙,溶于50mL三氯甲烷中,如不全溶,可用濾紙過(guò)濾于250mL分液漏斗中,用氨水溶液(1+99)提取三次,每次100mL,將提取液用棉花過(guò)濾至500mL分液漏斗中,用鹽酸溶液(1+1)調(diào)至酸性,將沉淀出的二硫腙用三氯甲烷提取2次~3次,每次20mL,合并三氯甲烷層,用等量水洗滌兩次,棄去洗滌液,在50℃水浴上蒸去三氯甲烷。精制的二硫腙置硫酸干燥器中,干燥備用。或?qū)⒊恋沓龅亩螂暧?00mL、200mL、100mL三氯甲烷提取三次,合并三氯甲烷層為二硫腙-三氯甲烷溶液。
17.2.8鹽酸羥胺溶液(200g/L):稱(chēng)20g鹽酸羥胺,加水溶解至50mL,加2滴酚紅指示液(1g/L),加氨水溶液(1+1),調(diào)pH至8.5~9.0(由黃變紅,再多加2滴),用二硫腙-三氯甲烷溶液(0.5g/L)提取至三氯甲烷層綠色不變?yōu)橹梗儆萌燃淄橄炊危瑮壢ト燃淄閷樱畬蛹欲}酸溶液(1+1)至呈酸性,加水至100mL,混勻。
17.2.9檸檬酸銨溶液(200g/L):稱(chēng)取50g檸檬酸銨,溶于100mL水中,加2滴酚紅指示液(1g/L),加氨水溶液(1+1),調(diào)pH至8.5~9.0,用二硫腙-三氯甲烷溶液(0.5g/L)提取數(shù)次,每次10mL~20mL,至三氯甲烷層綠色不變?yōu)橹梗瑮壢ト燃淄閷樱儆萌燃淄橄炊危看?mL,棄去三氯甲烷層,加水稀釋至250mL,混勻。
17.2.10氰化鉀溶液(100g/L):稱(chēng)取10g氰化鉀,用水溶解后稀釋至100mL,混勻。
17.2.11二硫腙使用液:吸取1.0mL二硫腙-三氯甲烷溶液(0.5g/L),加三氯甲烷至10mL,混勻。用1cm比色杯,以三氯甲烷調(diào)節(jié)零點(diǎn),于波長(zhǎng)510nm處測(cè)吸光度(A),用式(3)算出配制100mL二硫腙使用液(70%透光率)所需二硫腙-三氯甲烷溶液(0.5g/L)的毫升數(shù)(V)。量取計(jì)算所得體積的二硫腙三氯甲烷溶液,用三氯甲烷稀釋至100mL。
V=10×(2-lg70)/A=1.55/A…………………………(3)
17.3標(biāo)準(zhǔn)品
硝酸鉛[Pb(NO3)2,CAS號(hào):10099-74-8]:純度>99.99%。或經(jīng)國(guó)家認(rèn)證并授予標(biāo)準(zhǔn)物質(zhì)證書(shū)的一定濃度的鉛標(biāo)準(zhǔn)溶液。
17.4標(biāo)準(zhǔn)溶液配制
同10.4。
18、儀器和設(shè)備
注:所有玻璃器皿均需硝酸(1+5)浸泡過(guò)夜,用自來(lái)水反復(fù)沖洗,最后用水沖洗干凈。
18.1分光光度計(jì)。
18.2分析天平:感量0.1mg和1mg。
18.3可調(diào)式電熱爐。
18.4可調(diào)式電熱板。
19、分析步驟
19.1試樣制備
同5.1。
19.2試樣前處理
同5.2.1。
19.3測(cè)定
19.3.1儀器參考條件
根據(jù)各自?xún)x器性能調(diào)至最佳狀態(tài)。測(cè)定波長(zhǎng):510nm。
19.3.2標(biāo)準(zhǔn)曲線的制作
吸取0mL、0.100mL、0.200mL、0.300mL、0.400mL和0.500mL鉛標(biāo)準(zhǔn)使用液(相當(dāng)0μg、1.00μg、2.00μg、3.00μg、4.00μg和5.00μg鉛)分別置于125mL分液漏斗中,各加硝酸溶液(5+95)至20mL。再各加2mL檸檬酸銨溶液(200g/L),1mL鹽酸羥胺溶液(200g/L)和2滴酚紅指示液(1g/L),用氨水溶液(1+1)調(diào)至紅色,再各加2mL氰化鉀溶液(100g/L),混勻。各加5mL二硫腙使用液,劇烈振搖1min,靜置分層后,三氯甲烷層經(jīng)脫脂棉濾入1cm比色杯中,以三氯甲烷調(diào)節(jié)零點(diǎn)于波長(zhǎng)510nm處測(cè)吸光度,以鉛的質(zhì)量為橫坐標(biāo),吸光度值為縱坐標(biāo),制作標(biāo)準(zhǔn)曲線。
19.3.3試樣溶液的測(cè)定
將試樣溶液及空白溶液分別置于125mL分液漏斗中,各加硝酸溶液至20mL。于消解液及試劑空白液中各加2mL檸檬酸銨溶液(200g/L),1mL鹽酸羥胺溶液(200g/L)和2滴酚紅指示液(1g/L),用氨水溶液(1+1)調(diào)至紅色,再各加2mL氰化鉀溶液(100g/L),混勻。各加5mL二硫腙使用液,劇烈振搖1min,靜置分層后,三氯甲烷層經(jīng)脫脂棉濾入1cm比色杯中,于波長(zhǎng)510nm處測(cè)吸光度,與標(biāo)準(zhǔn)系列比較定量。
20、分析結(jié)果的表述
同13。
21、精密度
在重復(fù)性條件下獲得的兩次獨(dú)立測(cè)定結(jié)果的絕對(duì)差值不得超過(guò)算術(shù)平均值的10%。
22、其他
以稱(chēng)樣量0.5g(或0.5mL)計(jì)算,方法的檢出限為1mg/kg(或1mg/L),定量限為3mg/kg(或3mg/L)。


掃描二維碼
版權(quán)所有©廣州格丹納儀器有限公司 ![]() 粵公網(wǎng)安備 44011202000628號(hào) 粵ICP備14047970號(hào)
粵公網(wǎng)安備 44011202000628號(hào) 粵ICP備14047970號(hào)
分享到:







